Fenilketonurija - klasični znakovi, kao nasljedna i dijetalna terapija
Sadržaj
- 1Kako se manifestira bolest fenilketonurije
- 2Mehanizam razvoja bolesti
- 3Fenilketonurija u djece
- 4Simptomi bolesti
- 5Uzroci i provokativni čimbenici
- 6Dijagnostika
- 7Liječenje klasične fenilketonurije
- 8Hranidba novorođenčadi i dijetalna terapija
- 9Dijeta za predškolsku djecu i školsku djecu
- 10Skupine proizvoda na PKU
- 11Kako kontrolirati razinu fenilalanina u krvi
- 12
Bolest, čija je pojava povezana s defektima genetskog staničnog aparata - fenilketonurija - uključena je u mali popis nasljednih bolesti koje se liječe. Otkrivač ove bolesti bio je norveški liječnik IA Felling, a kasnije je otkriveno da razvoj i tijek bolesti odgovara jednom genu koji se zove gen fenilalanin hidroksilaze (dugi rameni 12. kromosoma koji sadrži do 4,5% ukupnog DNK staničnog materijala). Naslijeđeni nedostatak dovodi do djelomične ili potpune deaktivacije enzima jetre fenilalanin-4-hidroksilaze.
Kako se manifestira bolest fenilketonurije

Mehanizam razvoja bolesti
Inhibicija metabolita uzrokovana mutacijskom inaktivacijom enzima je aktivacija pomoćnih putova za razmjenu fenilalanina. Aromatska alfa-aminokiselina, kao rezultat neispravnih procesa razmjene, raspada se u toksične derivate koji se ne formiraju u normalnim uvjetima:
- fenilpiruvična kiselina (fenilpiruvat) - masna aromatska alfa-ketoacid, njeno obrazovanjedovodi do mijelinizacije procesa neurona i demencije;
- fenilmaktična kiselina - proizvod nastao tijekom regeneracije fenil virionske kiseline;
- Feniletilamin - početni spoj za biološki aktivne odašiljače elektrokemijskih impulsa, povećava koncentraciju dopamina, adrenalina i norepinefrina;
- Orthofhenylacetate - otrovna tvar koja uzrokuje kršenje metaboličkih procesa fekalnih spojeva u mozgu.
Medicinska statistika pokazuje da je patološki izmijenjeni gen prisutan u 2% populacije, ali se ne manifestira. Genetski defekt se prenosi na dijete od roditelja samo u prisutnosti bolesti kod oba partnera, a dijete u 50% slučajeva postaje nositelj mutiranog gena, ostajući zdrav. Vjerojatnost da će fenilketonurija kod novorođenčadi dovesti do bolesti je 25%.
Po kojoj se vrsti nasljeđuje
Fenilketonurija u djece
PKU tip II karakterizira činjenica da se prvi klinički simptomi javljaju nakon 1,5 godine od trenutka početka svjetlosti. Znaci bolesti ne nestaju nakon dijagnosticiranja genetskih abnormalnosti i početka terapije. Ova vrsta urođenih bolesti često dovodi do fatalnog ishoda 2-3 godine života djeteta. Najčešći simptomi tipa II PKU su:
- izražena odstupanja u mentalnom razvoju;
- hiperrefleksija;
- kršenje motoričkih funkcija svih ekstremiteta;
- sindrom nekontroliranih kontrakcija mišića.
- visok stupanj mentalne retardacije;
- prividno smanjenu veličinu lubanje u odnosu na druge dijelove tijela;
- spastičnost mišića (uz mogućnost potpunog gubitka pokreta ekstremiteta).

Manifestacije oborene bolesti
Tijekom kliničkih ispitivanja i opažanja, sugerirano je da je učinak biotoksični derivati fenilalanina razmjene uzrokuju smanjenje intelektualnih sposobnosti, što je progresivno i može dovesti do demencije (oligofrenija, idioti). Među vjerojatnim uzrocima ireverzibilnih povreda moždane aktivnosti smatra se da je najrazumnija posljedica smanjenja razine tirozina, nedostatka neurotransmitera koji prenose impulse između neurona.
Točna uzročno-posljedična veza između nasljednih bolesti i poremećaja mozga do sada nije otkrivena, kao i mehanizam razvoja zbog fenilketonurije takvih mentalnih stanja kao što su ehpraksija, eholalija, divlji napadi i razdražljivost. Rezultati analiza pokazuju da fenilalanin ima izravan toksični učinak na mozak, što također može uzrokovati smanjenje inteligencije.Osobitosti strukture i fenotipa
S obzirom da zasićenost pigmenta kože i kose ovisi o razini tirozina u mitohondrijima hepatocita, a fenilketonurija dovodi do zaustavljanja konverzije fenilalanina, bolesnici s ovom bolešću imaju fenotipske značajke (recesivni znakovi). Povećani tonus mišića uzrokuje pojavu odstupanja u strukturi tijela - postaje displastična. Izvrsna vanjska svojstva fenilketonurija uključuju:
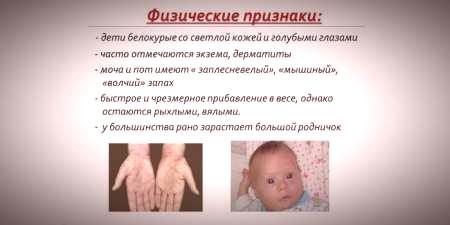
- hipopigmentacija - svijetla koža, blijedo plave oči, bezbojna kosa;
- cinizam udova;
- smanjena veličina glave;
- specifičan položaj tijela - kada pokušavate stajati ili sjediti dijete zauzima položaj "krojača" (ruke i noge savijene u zglobovima).
Simptomibolest
Pravodobno, Fellingova se bolest uspješno liječi prilagodbom prehrane, a razvoj djeteta odvija se u skladu s njegovom dobnom skupinom. Poteškoća u otkrivanju mutacije gena leži u činjenici da je rane znakove teško otkriti čak i iskusnom pedijatru. Ozbiljnost simptoma kongenitalne bolesti raste kako dijete raste, jer uporaba proteinske hrane doprinosi razvoju CNS poremećaja.
Znakovi u novorođenčadi
Tijekom prvih dana života djeteta, teško je otkriti znakove patoloških abnormalnosti - dijete se ponaša prirodno, nema kašnjenja u razvoju. Simptomi bolesti pojavljuju se za 2-6 mjeseci nakon rođenja. Roditelji bi trebali biti oprezni s ponašanjem djeteta koje karakterizira niska aktivnost, letargija ili, s druge strane, tjeskoba, hiperbolljivost.
Početkom dojenja novi proteini počinju ulaziti u tijelo novorođenčadi s mlijekom, što služi kao katalizator za pojavu prvih znakova, što jasno ukazuje na to da je bolest počela napredovati. Specifične kliničke manifestacije bolesti uključuju:- trajno povraćanje (često za urođeno sužavanje golmana);
- česta dislokacija;
- nema reakcije na vanjske podražaje;
- mišićna distonija (smanjena napetost mišića);
- konvulzivni sindrom (epileptički ili neepileptički napadaji).
Simptomi u djece nakon 6 mjeseci
Ako manifestacija genetske bolesti nijese dogodilo (ili nije viđeno) tijekom prvih 6 mjeseci od rođenja djeteta, a nakon tog razdoblja već možete točno odrediti zaostatak psihomotornog razvoja. Simptomi genetskih poremećaja uzrokovanih nedostatkom enzima kod djece starije od šest mjeseci su:
- smanjenje aktivnosti (do potpune indiferentnosti);
- nedostatak pokušaja ustajanja, sjedenja;
- poseban "mišji" miris kože (miris plijesni nastaje zbog povlačenja toksičnih derivata fenilalanina preko žlijezda znoja i urina);
- gubitak sposobnosti vizualizacije lica roditelja;
- ljuštenje kože;
- pojava dermatitisa, ekcema, skleroderme.
Progresija bolesti u odsustvu liječenja u djetinjstvu
- mikrocefalija (smanjena veličina mozga);
- otjecanje (pomicanje gornjeg retka zuba prema naprijed);
- kasnije erupcije zuba;
- hipoplazija cakline (mršavost ili potpuno odsustvo zubne cakline);
- kašnjenje u jezičnom razvoju sve do potpunog nedostatka govora;
- 3, 4 stupanj oligofrenije (mentalna retardacija, mentalna retardacija);
- prirođena srčana bolest (defekti u strukturi srčanog mišića, srce,velike posude);
- poremećaji vegetativnog sustava (akrocijanoza, pojačano znojenje, arterijska hipotenzija);
- konstipacija.
Uzroci i provokativni čimbenici
za očitovanje o mutacija autosomno recesivno nasljeđivanje znakova defektnog gena mora biti naslijeđena od oba roditelja. Genetske bolesti ovog tipa nalaze se na istoj učestalosti kod novorođenčadi i djevojčica. Patogeneza PKU uzrokovana je kršenjem izmjene fenilalanina, koji se može pojaviti u 3 oblika. Liječenje dijetalnom terapijom podliježe klasičnoj fenilketonuriji tipa I. \ t
Atipični oblici bolesti mogu se izliječiti prilagodbom prehrane. Ta odstupanja su uzrokovane nedostatkom tetrahydropteryna, dehydropterynreduktazы (rijetko - pyruvoyltetrahydropterynsyntazы, gvanozin-5-tryfosfattsyklohydrolazы itd). Većina slučajeva fatalnih slučajeva zabilježena je kod pacijenata s rijetkim varijacijama u PKU, s kliničkim manifestacijama svih oblika bolesti sličnih. Rizik rađanja s mutiranim genom fenilalanin hidroksilaze povećava se ako su roditelji bliski srodnici (s usko povezanim brakovima).Dijagnostika
Sumnja na genetske bolesti dijagnosticiran na temelju skupnih podataka dobivenih od proučavanja povijesti bolesti - genealoškim podacima, rezultatima kliničkih, medicinske i genetskih istraživanja. Za pravovremeno otkrivanje urođenih bolesti (PKU, cistična fibroza, galaktosemija, itd.) Program obvezne maseLaboratorijski pregled svih novorođenčadi (neonatalni probir).
Ako su budući roditelji svjesni prisutnosti mutiranog gena, moderna medicina nudi načine otkrivanja defekta tijekom trudnoće (prenatalna dijagnoza fetusa invazivnom metodom). Za podjelu fenilketonurije prema vrsti ozbiljnosti koristi se uvjetna klasifikacija na temelju razine fenilalanina u fibrinogenoj tekućini dobivenoj iz plazma plazme:-
Teška fenilketonurija - 1200 mkmol /l.
- Prosječno - 60-1200 μmol /l.
- Jednostavno (nije potrebno liječenje) - 480 μmol /l.
Test probira

Ako se ne otkriju patološke promjene, dijagnoza završava, što se bilježi u djetetovoj iskaznici. U slučaju odstupanja od norme, rezultati dijagnoze šalju se liječniku-pedijatru kako bi se omogućilo preciznije ispitivanje uzorka krvi novorođenčeta. Zdravlje djeteta ovisi o pravovremenom i točnom izvršavanju svih mjera za otkrivanje abnormalnosti. Ako se dijagnoza potvrdi nakon ponovljenog probirnog testa, roditelji djeteta će to učinitiu kliniku za dječju genetiku radi liječenja.
Analize i studije za potvrdu dijagnoze
Ponovna dijagnoza u slučaju otkrivanja odstupanja od norme tijekom početnog probirnog testa provodi se ponovnom procjenom analiza. Osim određivanja sadržaja fenilalanina u krvi na metode dijagnoze PKU u djece i odraslih uključuju:
- Ispitivanje sječe - određivanje fenilpiruvične kiseline u mokraći dodavanjem željeznog klorida u biomaterijal (postoji plavo-zelena obojenost);
- Gatree test - procjena stupnja reakcije mikroorganizama na metaboličke produkte ili enzime koji se nalaze u krvi pacijenta;
- kromatografija - proučavanje kemijskih svojstava tvari raspodijeljenih između dvije faze;
- fluorimetrija - ozračivanje biomaterijala monokromatskim zračenjem kako bi se odredila koncentracija tvari koje se u njoj nalaze;
- elektroencefalografija - dijagnoza električne aktivnosti mozga;
- snimanje magnetskom rezonancijom - kršenje atomskih jezgri stanica elektromagnetskim valovima i mjerenje njihovog odgovora.
Liječenje klasične fenilketonurije
- kod novorođenčadi i djece mlađe od 3 godine - do 242 μmol /l;
- za predškolsku djecu - do 360 mkmol /l;
- u bolesnika u dobi od 7 do 14 godina - do 480 μmol /L;
- u adolescenata - do 600 μmol /l.

- proteinski hidrolizati (amigeni, aminazol, fibrinoze);
- ne sadrže smjese fenilalanina zasićene esencijalnim aminokiselinama - Tetrafen, fenil slobodan.
Značajke liječenja atipičnih oblika
- tetrahidrobiopterin - faktor utjecaja enzima;
- sintetski analozi tetrahidrobiopterina - te tvari bolje prodiru kroz krvno-moždanu barijeru;
- lijekovi za supstitucijsku terapiju - ne uklanjaju uzrok fenilketonurije, već podupiru normalno funkcioniranje tijela (levodopa zajedno s karbidopom, 5-oksitriptofanom, 5-formiltetrahidrofolatom);
- hepatoprotektori - podržavaju funkcioniranje jetre;
- antikonvulzivi;
- uvođenje gena fenilalanin hidroksilaze u jetru - eksperimentalna metoda.
Značajke prehrane novorođenčadi i terapija prehrane
U prvoj godini života djeteta dopušteno je dojiti, ali treba ograničiti njezinu količinu. Do 6 mjeseci, prihvatljiva razina potrošnje fenilalanina je 60-90 mg na 1 kg tjelesne težine bebe (u 100 g mlijeka sadrži 5,6 mg fenilalanina). Počevši od 3 mjeseca, dijetalna dijeta treba postupno povećavati uvođenjem voćnih sokova i pirea.
Za djecu od 6 mjeseci dopušteno je unošenje u prehranu biljnog pirea, kaše (od sagoa), bez proteinaKiselev. Nakon 7 mjeseci bebi možete dati tjesteninu s niskim sadržajem proteina, s 8 mjeseci - kruh bez proteina. Nije ustanovljena starost u kojoj treba ograničiti prijenos proteina u tijelu bolesnog djeteta. Liječnici i dalje raspravljaju o svrsishodnosti dijetetske terapije, ali se slažu da bi se trebala poštivati najmanje 18 godina, prehrambena prehrana.
Fenilketonurija, dijagnosticirana sa ženom, nije razlog za napuštanje rođenja djeteta. Buduće majke s PKU kako bi spriječile oštećenje fetusa tijekom trudnoće i spriječile moguće komplikacije potrebne su prije planiranog začeća i tijekom nošenja djeteta da se pridržavaju prehrane s ograničenjem fenilalanina (razina u krvi treba biti do 242 mikromola /l).Mješavine cjepiva za bebe
- za bebe do godine imenovane Affenilak 15, Analog-JV, PKU-1, PKU-mix, PKU Anamix;
- za djecu stariju od 1 godinegodine, propisuju obogaćene vitaminima i mineralima mješavinu visokog sadržaja proteina - PKU Prima, P-AM Universal, PKU-1, PKU-2, HRM Maxamed, HR Maxumum.

Prehrambeni proizvodi za nadopunu zaliha proteina
- Berlofen;
- Tsimorgan;
- Minafen; Apotoni.
Dijeta za predškolsku djecu i školsku djecu
- procjena neuroloških parametara, psihološkog stanja;
- kontrola parametara elektroencefalograma;
- određivanje razine fenilalanina.
Skupine proizvoda na PKU
Crveni popis
- meso;
- unutarnji organi životinja, nusproizvodi;
- kobasice, kobasice;
- plodovi mora (uključujući ribe);
- jaja svih ptica;
- kiselo mliječni proizvodi; Matice;
- mahunarke i žitarice;
- proizvodi od soje;
- želatinozna posuda;
- slastice;
- aspartam.
Narančasti popis
- konzervirano povrće;
- jela od krumpira i riže;
- kupus;
- mlijeko;
- šerbet.
Zeleni popis
- voće;
- povrće (osim krumpira i kupusa);
- bobice;
- zelje;
- škrobaste žitarice (sago);
- med, šećer, džem;
- brašno od kukuruznog ili rižinog brašna;
- maslac, masnoće (maslac, suncokret, maslina).
Kako kontrolirati razinu fenilalanina u krvi
- do 3 mjeseca - pregled krvi treba provoditi tjedno dok se ne dobiju stabilni rezultati;
- od 3 mjeseca do 1 godine - 1-2 puta mjesečno;
- od 1 do 3 godine - 1 put u 2 mjeseca;
- preko 3 godine - tromjesečno.

Videozapisi
Informacije u članku su informativne. Materijali članka ne zahtijevaju neovisni tretman. Samo kvalificirani liječnik može dijagnosticirati i dati savjet o liječenju na temelju individualnih karakteristika određenog pacijenta.















